Drug-excipient compatibility studies represent an important phase in drug development. Before a drug substance is formulated into the desired dosage form, there is need for the formulation scientist to fully consider the chemical structure of the drug substance, the type of delivery system required and the proposed manufacturing process.
Drug substances are usually combined with excipients which serve different and specialized purpose. Although excipients are pharmacologically inert, they can undergo chemical reactions and physical interactions with drug substances under favourable environmental conditions. These interactions can lead to instability resulting in the formation of new entities with different physicochemical properties and pharmacological effects.
Drug-excipient compatibility studies have been used as an approach for accepting/rejecting excipients for use in pharmaceutical formulations, thus allowing the rapid optimization of a dosage form with respect to patentability, processing, drug release, elegance, and physicochemical stability.
In order to obtain rapid stability assessment of drug and excipients, drug stability is investigated under the stress condition according to standard protocol and/or existing knowledge on potential degradation pathway or incompatibilities.
Contents
Read also: Preformulation Studies: Stability analysis
The mechanisms of drug-excipient(s) interactions are not fully understood despite the best efforts of several eminent investigators in the field. However, some of the common ways by which excipients may alter drug stability in a dosage form include:
These types of interactions are quite common but are very difficult to detect in dosage forms. Drug substances and excipients interact without undergoing changes involving breaking or formation of new bonds.The components of the drug product retain their chemical structure but undergo changes which alter their physical properties.
Physical interactions may result in changes in dosage uniformity, colour, odour, flow properties, solubility, sedimentation rate, dissolution rate etc. Incompatibilities are assessed by physically observing the test samples.
Physical interactions can be either beneficial or detrimental to the product performance depending on its application.
This involves the interaction of drug substance and excipient through chemical degradation pathway. The formulation undergoes a chemical reaction in which the constituent atoms are rearranged via bond breakage and bond formation to produce an unstable chemical entity.
Generally, chemical interactions have a deleterious effect on the formulation hence; such kind of interactions must be avoided.
Chemical interactions can be in the form of hydrolysis, oxidation, racemization, polymerization, Maillard reactions, photolysis etc., and changes in the study samples are analyzed by a chromatographic-based assessment of potency and formation of degradants or by any other analytical method depending on the nature of the candidate drug molecule, available literature and the goals of the study.
Some examples of chemical drug-excipient interactions include
By this, we mean interactions that occur after the drug product has been administered to the patient. These interactions are similar to physical interactions but differ in the sense that
All excipients interact in a physiological sense when they are administered as part of a dosage form. They are included in a formulation specifically because they interact with the physiological fluids and function in certain ways e.g., disintegrants in immediate release tablets and capsule formulations. On the other hand, physiological interactions can be detrimental to the patient.
Examples of such interactions include:
Read Also: Preformulation Studies: Solubility analysis
The key to the early assessment of instability in formulations is the availability of analytical methods to detect low levels of degradation products, generally less than 2%. Below are some of the analytical methods which are used in drug-excipient compatibility studies.
Thermal methods of analysis comprise a group of techniques in which the physicochemical properties of drug substances are measured as a function of temperature. In this method, the test samples are subjected to a controlled temperature over a given period of time. This method of analyses plays a vital role in drug-excipient compatibility studies and has been frequently used for quick identification of physicochemical interaction between drugs and excipients.
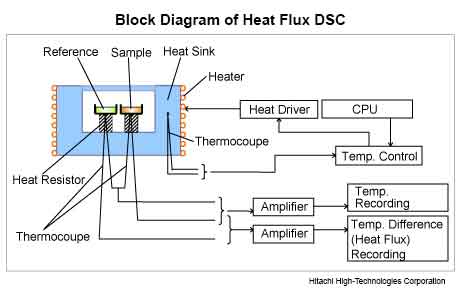
DSC represents a leading thermal screening technique that has been increasingly used for excipient compatibility studies for over five decades. In this technique, the DSC curves of pure samples are compared to that obtained from 50% mixture of the drug and excipient (usually 5mg of the drug in a ratio of 1:1 with the excipient).
It is assumed that the thermal properties (melting point, change in enthalpy, etc.) of blends are the sum of the individual components if the components are compatible with each other. An absence, a significant shift in the melting of the components or appearance of a new exo/endothermic peak and/or variation in the corresponding enthalpies of reaction in the physical mixture indicates incompatibility.
However, slight changes in peak shape height and width are expected due to possible differences in the mixture geometry.
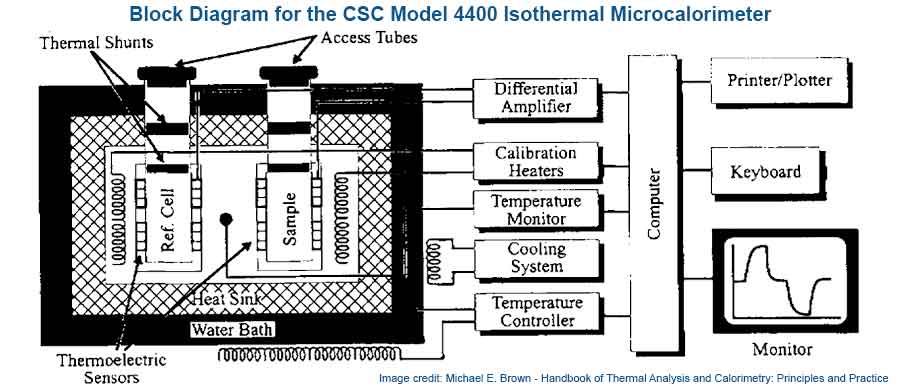
This is an extremely sensitive and invaluable tool used to determine drug-excipient incompatibilities. It measures minute amounts of heat emitted or absorbed by a sample in a variety of processes. This method of analysis is used to characterize pharmaceutical solid to obtain heats of solution, heats of crystallization, heats of reaction, heats of dilution and heats of adsorption – since nearly all physicochemical processes are accompanied by a heat exchange within their surroundings.
In a typical drug-excipient compatibility study, a solution, suspension, or solid mixture of drug substance and excipient is placed in the calorimeter and the thermal activity (heat gained or evolved) at a constant temperature is monitored.
The thermal activity observed is assumed to be proportional to the rate of chemical and/or physical processes taking place in the sample. The thermal activity of the test sample is compared to the “non-interaction” curve constructed from the control (i.e., thermal activity of drug substance and excipient that were measured individually). If an experimentally significant difference is observed, the excipient is considered to be potentially incompatible with the drug substance.
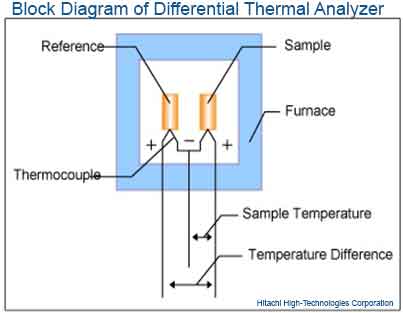
Differential Thermal Analysis (DTA) is an analytical technique in which the changes in temperature between a test sample and an inert reference under controlled and identical conditions is used to identify and quantitatively analyze the chemical composition of a substance.
When the test sample and inert reference are heated to a sufficient temperature, the thermal changes in the test sample which lead to the absorption or emission of heat can be detected relative to the inert reference (control). The differences in temperature are then plotted against time, or against temperature.
Drug-excipient interactions can be identified by comparing DTA curves obtained from the test sample with those of inert reference. Incompatibilities are indicated by the appearance of one or more new DTA peaks or the disappearance of one or more DTA peaks corresponding to those of the components of the test sample. In the absence of any interaction, the DTA peak of the test sample show patterns corresponding to those of the individual components.
Read Also: Preformulation Studies: Bulk Characterization
Spectroscopic analytical methods include all techniques which probe certain features of a given sample by measuring the amount of radiation emitted or absorbed by molecular or atomic species of interest.
This method of analysis uses electromagnetic radiation to interact with matter and thus investigate certain features of a sample as a function of wavelength (λ). Because these methods of analysis use a common set of optical devices for collimating and focusing the radiation, they often are identified as optical spectroscopies.
Some of the most frequently used spectroscopic methods of analysis include vibrational spectroscopy, diffuse reflectance spectroscopy, fluorescence spectroscopy, FT-IR spectroscopy etc. and each operates over different, limited frequency ranges within this broad-spectrum, depending on the processes and degree of the energy changes.
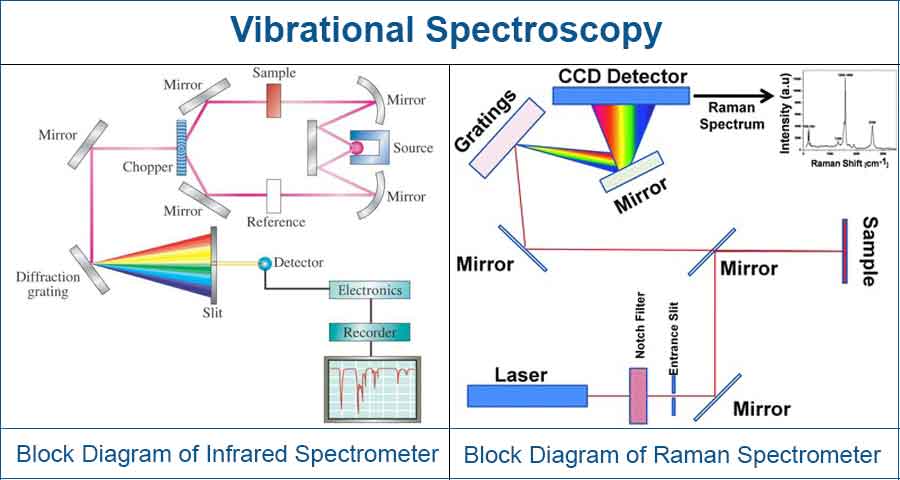
Using this method, information on the molecular structure and environment of organic compounds are generated by measuring the vibrations of chemical bonds that result from exposure to electromagnetic energy at various frequencies. These vibrations are commonly studied by infrared and Raman spectroscopies.
While infrared spectroscopy uses the infrared region of the electromagnetic spectrum (from about 400 cm-1 to 4000 cm-1) to measures the change in dipole moment, Raman spectroscopy uses inelastic scattering process to measures the change in polarization of the sample.
The spectra obtained are indicative of the nature of chemical bonds present in the test sample, and when pieced together can be used to identify the chemical structure or composition of a given sample.
Vibrational spectroscopy are not only used to investigate solid state properties of drug substances and their formulations, but are also used as compatibility study tool as the vibrational changes serve as probe of potential intermolecular interactions among the components. Thus, drug-excipient interactions that occur during processing can easily be detected with the aid of these spectroscopic techniques.
This is a type of spectroscopic techniques which analyzes fluorescence properties of samples in order to provide information regarding their concentration and molecular environments. It involves using a beam of light, usually UV/visible radiation, to excite the electrons in molecules of certain compounds particularly those with chromophore and rigid structure, causing them to emit the radiation at a longer wavelength. The radiation emitted (emission spectrum) and/or the radiation absorbed by the sample (excitation spectrum) can then be measured and compared with the control.
Apart from determining the stability of peptide drugs in solution, fluorescence spectroscopy has also been used in
Chromatography is an analytical technique frequently used in pharmaceutical research for separating sample mixture into its individual components. This technique is based on selective adsorption of the components on a stationary phase (usually a solid or liquid with high surface area).
As the solute mixture passes over the stationary phase, the components are adsorbed and released at the surface at varying rates depending on differential affinities of individual components towards stationary and mobile phase.
Compared to other available analytical techniques used in drug-excipient compatibility studies, chromatography is known for its characteristics of high resolution and detection power, making it suitable for detecting multiple components in a complex mixture with high accuracy, precision, specificity, and sensitivity. Various chromatographic methods of analysis have been used in drug-excipient compatibility studies, all following the same basic principles of operation.
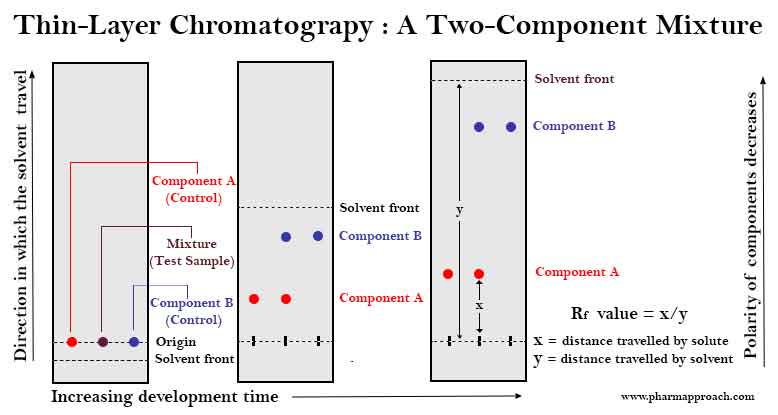
TLC is a chromatographic method of analysis carried out on glass, plastic or metal plates coated on one side with a thin layer of adsorbent. The thin layer of adsorbent serves as the stationary phase and is usually made of silica, alumina, polyamide, cellulose or ion exchange resin.
In TLC, solutions of the test samples (that is, a mixture of the drug and the excipient) and the controls (individual drug and excipients) are prepared and spotted on the same baseline at the end of the plate (the origin). The plate is then placed upright in a closed chamber containing mixtures of organic solvents which serve as the mobile phase. The analyte moves up the plate, under the influence of the mobile phase which moves through the stationary phase by capillary action.
The distance moved by the analyte is dependent on its relative affinity for the stationary or the mobile phase. Incompatibilities are indicated by the formation of a spot with Rf value (retardation factor) different from that of the controls after the plate has been developed with solvent.
An excipient on the other hand is considered to be potentially compatible with the drug substance if the spots produced have identical Rf value with those of the controls. Because some samples undergo negligible thermal changes which might be difficult to detect by thermal methods of analysis, TLC is widely used in drug-excipient compatibility study as a confirmative test of compatibility after performing DSC.
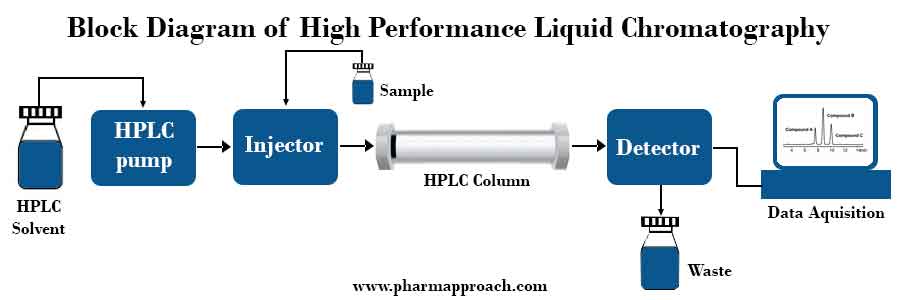
HPLC is a chromatographic technique widely used in drug-excipient compatibility studies by quantitative estimation of test samples that have been subjected to isothermal stress testing (IST). This method of analysis is based on mechanisms of adsorption, partition and ion exchange, depending on the nature of the stationary phase used.
In HPLC, a liquid mobile phase is pumped under high pressure through the stationary phase (a stainless-steel column packed with tiny particles with a diameter of 3 to 10 micron). A small volume of the test sample is loaded onto the head stainless-steel column via a loop valve.
Separation of a sample mixture occurs according to the relative lengths of time spent by its components in the stationary phase. Column effluent can be monitored with a variety of flow-through device/detector that measures the amount of the separated components.
HPLC results that show a percentage loss similar to the control (drug considered individually) indicate no interaction between drug and the excipients and vice versa.
Read Also: Preformulation Studies: A Foundation for Dosage Form Development
Drug-excipient compatibility study is a necessary prerequisite to the development of drug products that are safe and stable for use. Proper selection and assessment of possible incompatibilities between the drug and excipients during preformulation studies is of paramount importance to accomplish the target product profile and critical quality attributes.
In order to avoid stability problems encountered during drug development and post-commercialization, there is need for proper assessment of possible incompatibilities between the drug and excipients using appropriate analytical techniques. These analytical techniques are needed not only to generate useful information with regards to which excipient is compatible with a drug substance, but also for troubleshooting unexpected problems which might arise during formulation processes.
Drug-excipient interactions may take a long time to be manifested in conventional stability testing programs, and are not always predicted by stress and pre-formulation studies. It is hoped that this write-up provides valuable information concerning the drug–excipient interactions that aid in the selection of appropriate excipients for safe, stable and bioavailable dosage form.
Related keyword: drug-excipient compatibility studies slideshare, drug excipient compatibility studies in preformulation slideshare, drug-excipient compatibility studies guidelines fda, procedure for drug-excipient compatibility study, drug-excipient compatibility studies slide share, drug-excipient interaction methods, drug excipient interaction different methods ppt, drug-excipient interaction different methods pdf
Comments6







This article is very well written in a very simple language that the message is easily apprehended. I look forward to be reading more because I would love to work in the big pharma setting one day.
I am glad you found it useful.
Really good, indeed.
this is very insightful…
I am happy you found it useful.
Very elaborate narration and technical details. It also gives insight for estimation, advantages and disadvantages. It is in depth and very useful details for carrying out compatibility pharmaceutical formulation. It is great service to pharmacists and pharma personnel.