Tablets are solid drug delivery system prepared by compressing a single dose of one or more active drug substance(s) with some additives/ pharmaceutical excipients. They may be circular, oblong, oval, triangular or cylindrical in shape and flat-, round-, concave- or convex-faced with straight or bevelled edges.
In tablet formulation development and during manufacturing of tablet dosage forms, a number of quality control tests are performed to ensure that tablets produced meet the requirements as specified in official compendium and conventional requirements established by the industries over the years. These tests can be grouped into two broad categories namely:
Contents
They are called official tests because the test methods are described in official compendia such as the British Pharmacopoeia, American Pharmacopoeias etc. They are standardized test procedures which have clearly stated limits under which compressed tablets could be accepted. These tests include:
These tests are traditionally concerned with the content and the in vitro release of the active ingredient. It must be emphasized that what is presented here should by no means replace what are presented on each of the tests in official compendia.
Read Also: In-Process Quality Control (IPQC) of Pharmaceutical Dosage Forms
This is determined from a sample of 20 tablets which should be randomly selected from a batch of tablets. The tablets are weighed together and are crushed in a mortar with a pestle.
An amount equivalent to the theoretical content of each tablet or the average of the crushed tablets is weighed out in an analytical balance. The weighed powder is dispersed in a solvent in which the active drug is freely soluble or in a solvent prescribed in the individual drug monograph.
This is filtered and an aliquot of the resultant filtrate is subjected to the stipulated assay procedures. The assay procedures are usually given in the individual drug monograph.
Analysis of the active drug is usually carried out using spectrophotometry or High-Performance Liquid Chromatography (HPLC). The formulation scientist must be familiar with Beer-Lambert’s law. This could be found in relevant analytical textbooks.
It must be emphasized that the results obtained here gives the average content of 20 tablets but does not give indication of the variation of drug content among the individual tablets. The limits of acceptance or rejection of tablets batches are usually presented in the individual drug monograph.
The test for uniformity of weight is performed by weighing individually 20 tablets randomly selected from a tablet batch and determining their individual weights. The individual weights are compared with the average weight.
The sample complies with USP standard if no more than 2 tablets are outside the percentage limit and if no tablet differs by more than 2 times the percentage limit.
Coated tablets are exempted from these requirements but must conform to the test for content uniformity.
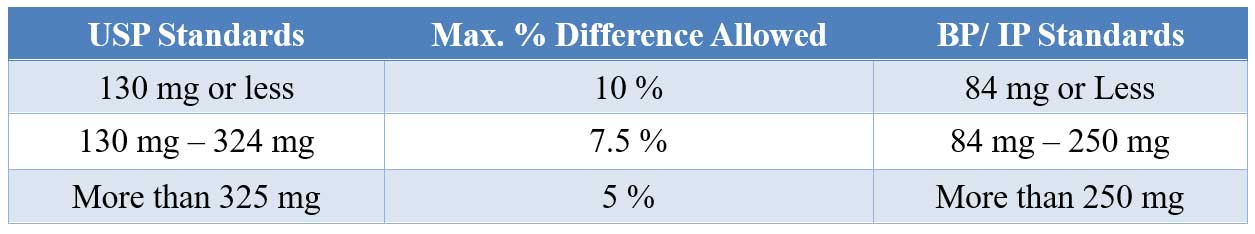
Weight variation tolerance for uncoated tablets
Uniformity of weight is a function of granulation quality, flow of granulation and machine performance. However, sometimes these ranges are not sufficiently narrow.
Read Also: Quality Control and Evaluation Parameters for Chewable Tablets
Content uniformity test was developed to ensure content consistency of active drug substances within a narrow range around the label claim in dosage units. This test is crucial for tablets having a drug content of less than 2 mg or when the active ingredient comprises less than 2% of the total tablet weight.
By the USP method, 30 tablets are randomly selected, 10 of these tablets are assayed individually according to the method described in the individual monograph. Unless otherwise stated in the monograph, the requirements for content uniformity are met if the amount of active ingredient in nine (9) of the ten (10) tablets lies within the range of 85% to 115% of the label claim. The tenth tablet may not contain less than 75% or more than 125% of the labelled drug content.
If one or more dosage units do not meet these criteria, the remaining 20 tablets are assayed individually and none may fall outside of the 85% to 115% range for the batch to be accepted.
Various factors are responsible for the variable content uniformity in tablets. This may include:
For tablets, the first important step towards drug dissolution is breakdown of the tablets into granules or primary powder particles, a process known as disintegration. All USP tablets must pass a test for disintegration, which is conducted in vitro using a disintegration test apparatus.

Disintegration test apparatus
The apparatus consists of a basket-rack assembly containing six open-ended transparent tubes of USP-specified dimensions, held vertically upon a 10-mesh stainless steel wire screen.
During testing, a tablet is placed in each of the six tubes of the basket, and through the use of a mechanical device, the basket is raised and lowered in a bath of fluid (e.g. water, or as prescribed in the individual drug monograph) at 29 to 32 cycles per minute, the wire screen always below the level of the fluid. For most normal release tablets, the time permitted is 15 minutes.
Tablets are said to have disintegrated if no fragments (other than fragments of coating) remains on the screen, or if particles remain, they are soft without an unwetted core. Chewable tablets are not required to comply with the test.
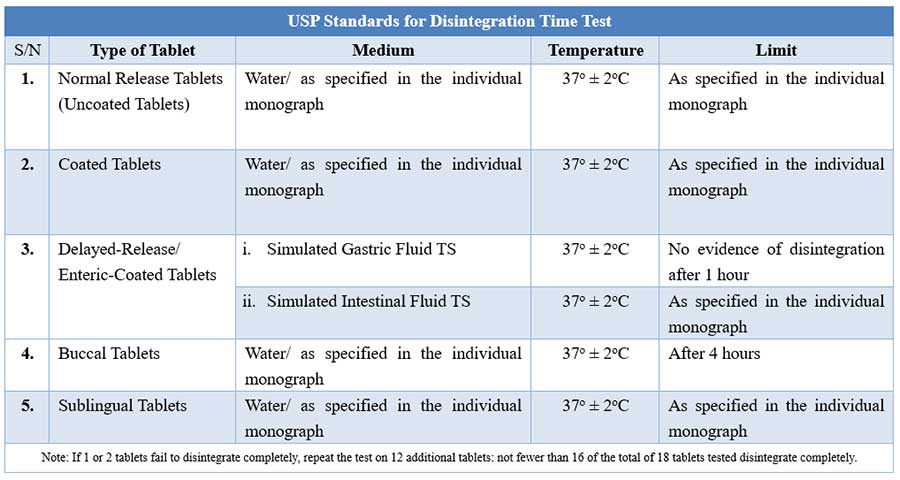
USP Disintegration testing condition and interpretations
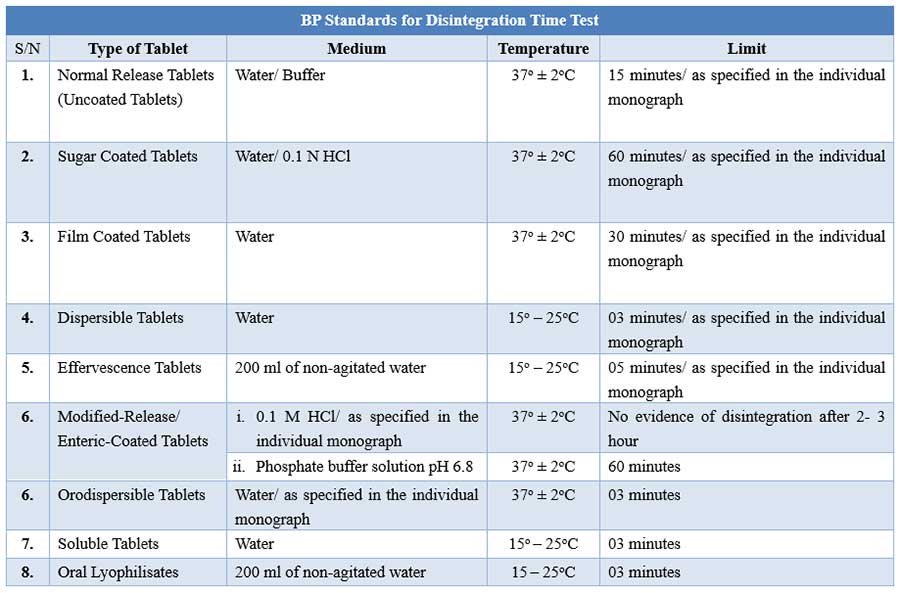
BP Disintegration testing condition and interpretations
Research has established that one should not automatically expect a correlation between disintegration and dissolution. However, since the dissolution of drug from the fragmented tablet appears to control partially or completely the appearance the drug in the systemic circulation, disintegration is still used as a guide by the formulator in the preparation of an optimum tablet formula and as an in-process control test to ensure batch to batch uniformity.
Factors affecting disintegration of tablets include:
To continue reading, click on the page buttons below…
Comments29






very informative…thanks
You are welcome. I am happy you found it useful.
Your Comment *I just get a lot of lessons from these and thank you very much!!!
You are welcome. I am happy you found it useful.
the piece has really helped me. please sir, how will i quote this as a reference in my research work
I am happy you found it useful. You can reference the webpage as shown below:
Ozioko, C. (2019, January 19). Quality Control Tests for Tablets [Web log post]. Retrieved June 8, 2019, from https://www.pharmapproach.com.
The content is informative and precise.
I am happy you found it useful.
I am so impressed with your posts and website. Thank you for such incredible and valuable information. I’ve learned and/or refresh my knowledge so much from it. You are truly appreciated.
Thanks for the compliment. I am happy you found it useful.
The sentences “A force of about 4 kg is considered the minimum requirement for a satisfactory tablet. Measurement is usually carried out using a minimum of ten tablets.” was gotten from Ansel’s Pharmaceutical Dosage Forms and Drug Delivery Systems (10th edition) by Allen L. V and Ansel H. C. (2014) page 272.
thanks very informative
You are welcome. I am happy you found it useful.
Have you any results on Indian herbal remedy for lowering blood pressure
Mukta Vati. Please?
No, we do not have any result on Indian herbal remedy for lowering blood pressure.
Thank you for your very helpful explanation.
You are welcome. I am happy you found it useful.
Thank you for this thorough and detailed explanation. It was very helpful.
You are welcome. I am happy you found it useful.
Thank you for this article, it has been very helpful
I am happy you found it useful.
This article is very interesting which is worth reading it . Thanks for information
Thanks
perfectly designed information
Thanks
Thank you so much. I want to ask, about the Disintegration test, must all the six disintegrate before you check the time?
Yes
The content is very informative and helpful. Thanks
You’re welcome