Contents
This test measures the amount of time required for a given percentage of the drug substance in a tablet to go into solution under a specified set of conditions. It is intended to provide a step toward the evaluation of the physiological availability of the drug substances.
In vitro dissolution test is performed using a variety of equipment/apparatus. The British Pharmacopoeia recommends three types of apparatus – the rotating basket, the rotating paddle and the flow-through cell. The static-basket magnetic stirrer assembly can also be used for this test.
The rotating paddle method is generally more discriminatory than the basket method. The flow-through cell method is very useful particularly for

USP Dissolution Apparatus 2
The dissolution medium for each drug is available in the individual drug monograph. For basic drugs, acidic media are used (e.g. 0.1 M hydrochloric acid) while alkaline media are used for acidic drugs (e.g. alkaline buffers). For drugs with non-ionizing molecules, water is recommended.
Dissolution rate test is performed at 37 ± 1 oC. Samples are removed from the dissolution chamber at periodic intervals and analyzed for drug content using a spectrophotometer. Dissolution samples removed for assay should be filtered to remove particles of drugs present, and to exclude tablet excipients that might otherwise interfere with the assay. Non-absorbent filter papers are recommended.
Most commonly, the results of dissolution tests are expressed in terms of the time required to release some percentage of labelled amount of drug from the dosage form. This approach is reported to be particularly useful for quality control purposes once the dissolution characteristics of a drug and dosage form are well understood.
For tablet dosage form design purposes, and for critical product comparison, however, the time required for substantially complete 80 to 90% release or amount released versus time profiles are the most desired approach.
While in vitro dissolution experiment may not correlate perfectly with in vivo bioavailability, the concept of dissolution efficiency proposed by Kahn and Rhodes could be employed to assess the most probable in vivo performance of a tablet formulation.
Dissolution test is not designed to measure the efficacy or safety of the tablet being tested. Both the effectiveness and safety of a specific dosage form must be demonstrated, initially, by means of appropriate in vivo studies and clinical evaluation.
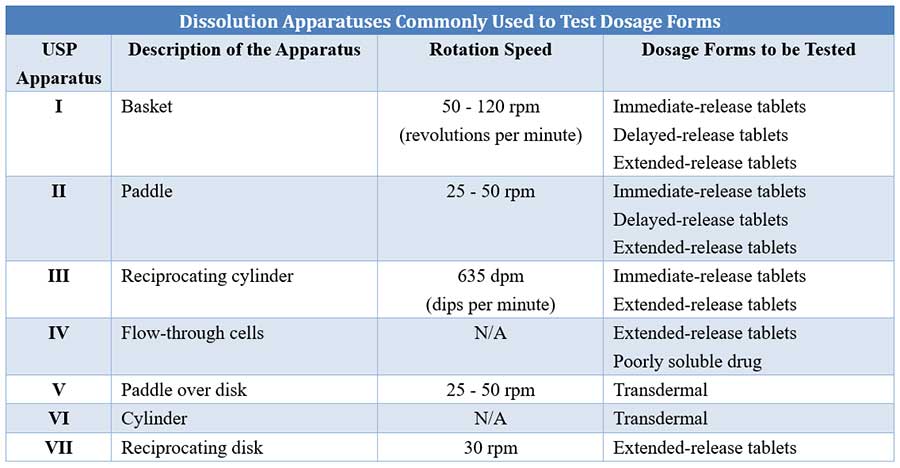
Comparison of different USP dissolution apparatuses
Various factors can affect the dissolution of a drug; they are classified under three categories as follows:
1. Physiochemical properties of the drug
2. Factors related to tablet manufacturing
3. Factors related to method of dissolution study
Read Also: Quality Control Requirements for Pharmaceutical Dosage Forms
These are tests that are performed on tablets and which are not listed in official compendia and concern a variety of quality attributes that need to be evaluated, such as the porosity of tablets, hardness or crushing strength test, friability test, tensile strength determination, thickness test etc.
Some of these tests have no officially set limits for acceptance or rejection and thus may vary from manufacturer to manufacturer and from formulation to formulation.
Crushing strength and friability appeared in the 2001 Edition of British Pharmacopoeia (Appendix A324). There was however no definite set limits. The two tests are, therefore, here considered under non-pharmacopoeial tests.
This measures the degree of force (in kilograms, pounds, or in arbitrary units) needed to fracture a tablet. Besides the concentration of binders used and the compression force, the hardness of a tablet depends on
The crushing strength of tablets is usually checked using Monsanto or Stokes hardness tester, Strong-Cobb hardness tester and the Pfizer crushing strength tester. All are manually used. So, strain rate depends on the operator.

Hardness Tester
Currently, electrically driven hardness testers such as those manufactured by SOTAX, Key, Van Kel, Erweka, Dr Schleuniger Pharmatron etc., are widely used to measure crushing strength of tablets.
These equipment eliminate the operator variability encountered with manual hardness testers. Newer equipment with printers are also available.
A force of about 4 kg is considered the minimum requirement for a satisfactory tablet. Measurement is usually carried out using a minimum of ten tablets.
It has been found that a linear relationship exists between crushing strength and the logarithm of compressional force, except at high forces.
The strength of a cylindrical flat-faced tablet can be expressed as a tensile strength (Ts). This can be calculated as follows:
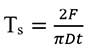
Where F is the force needed to fracture a cylindrical flat-faced tablet of thickness t along its diameter D.
This measures the resistance of tablets or granules to abrasion or fracture. The idea behind this test is to mimic the kind of forces, caused by phenomena such as collisions and sliding of tablets towards each other, which a tablet is subjected to during coating, packaging, handling, and shipping.
A minimum of 20 tablets are dedusted, weighed and subjected to a uniform tumbling motion for a specified time. They are then dedusted and reweighed.
The measure of abrasion/ friability loss is usually expressed as percentage loss in weight. It is calculated from the equation:
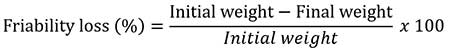
The test is rejected if any tablet caps, laminates or breaks up in course of the test. As a rule of thumb, a maximum weight loss of not more than 1% generally is considered acceptable for most pharmaceutical products. Values of up to 2% or above have been reported in direct compression formulations.
The friability of tablets may be influenced by moisture content. Chewable tablets show a high friability weight loss compared to conventional compressed tablets. A number of instruments are available for friability tests but the most popular and most reliable is the Roche Friabilator.

Dual drum friability tester
Tablet thickness is determined by the diameter of the die, the amount of fill permitted to enter the die cavity, the compaction characteristics of the fill material, and the force or pressure applied during compression. To manufacture tablets of uniform thickness during and between batch productions for the same formulation, care must be exercised to employ the same factors of fill, die, and pressure.
The degree of pressure affects not only thickness but also hardness of the tablet; hardness is perhaps the more important criterion since it can affect disintegration and dissolution. Thus, for tablets of uniform thickness and hardness, it is doubly important to control pressure. Tablet thickness also becomes an important characteristic in packing operations and in counting of tablets using filling equipment which uses the uniform thickness of the tablets as a counting mechanism.
Read Also: Quality Control Tests for Capsule Drug Products
Tablet thickness is measured with a vernier caliper, thickness gauge or automated equipment (Automatic weight, hardness, thickness, and tablet diameter test instrument). The thickness of a tablet should be controlled within ±5% variation of a standard value depending on the size of the tablet.
Other non-pharmacopoeial tests include measurement of tablet diameter, porosity, liquid penetration, mechanical strength, and density.
Quality control of tablets involves various tests which require keen attention. To ensure that established product quality standards are met, these tests must be performed during production (in-process controls) and verified after the production of each batch.
Related keywords for Quality Control Tests for Tablets: quality control test for tablets slideshare, quality control tests for capsules, tablet hardness test limits, quality control test for capsules slideshare, Quality Control Tests for Tablets, weight variation test for tablets, quality control test for tablets as per usp, tablet evaluation tests pdf, finished product testing of tablets, Quality Control Tests for Tablets, in-process quality control of tablets pdf, tablet hardness test acceptance criteria, weight variation test for tablets, ipqc test for tablets slideshare
Comments29






very informative…thanks
You are welcome. I am happy you found it useful.
Your Comment *I just get a lot of lessons from these and thank you very much!!!
You are welcome. I am happy you found it useful.
the piece has really helped me. please sir, how will i quote this as a reference in my research work
I am happy you found it useful. You can reference the webpage as shown below:
Ozioko, C. (2019, January 19). Quality Control Tests for Tablets [Web log post]. Retrieved June 8, 2019, from https://www.pharmapproach.com.
The content is informative and precise.
I am happy you found it useful.
I am so impressed with your posts and website. Thank you for such incredible and valuable information. I’ve learned and/or refresh my knowledge so much from it. You are truly appreciated.
Thanks for the compliment. I am happy you found it useful.
The sentences “A force of about 4 kg is considered the minimum requirement for a satisfactory tablet. Measurement is usually carried out using a minimum of ten tablets.” was gotten from Ansel’s Pharmaceutical Dosage Forms and Drug Delivery Systems (10th edition) by Allen L. V and Ansel H. C. (2014) page 272.
thanks very informative
You are welcome. I am happy you found it useful.
Have you any results on Indian herbal remedy for lowering blood pressure
Mukta Vati. Please?
No, we do not have any result on Indian herbal remedy for lowering blood pressure.
Thank you for your very helpful explanation.
You are welcome. I am happy you found it useful.
Thank you for this thorough and detailed explanation. It was very helpful.
You are welcome. I am happy you found it useful.
Thank you for this article, it has been very helpful
I am happy you found it useful.
This article is very interesting which is worth reading it . Thanks for information
Thanks
perfectly designed information
Thanks
Thank you so much. I want to ask, about the Disintegration test, must all the six disintegrate before you check the time?
Yes
The content is very informative and helpful. Thanks
You’re welcome